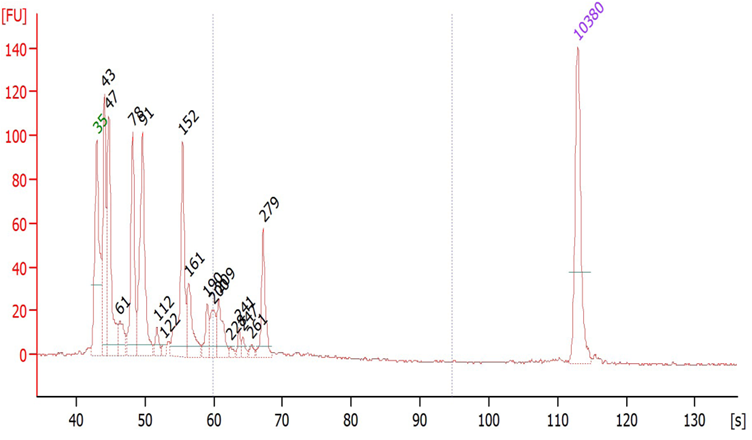
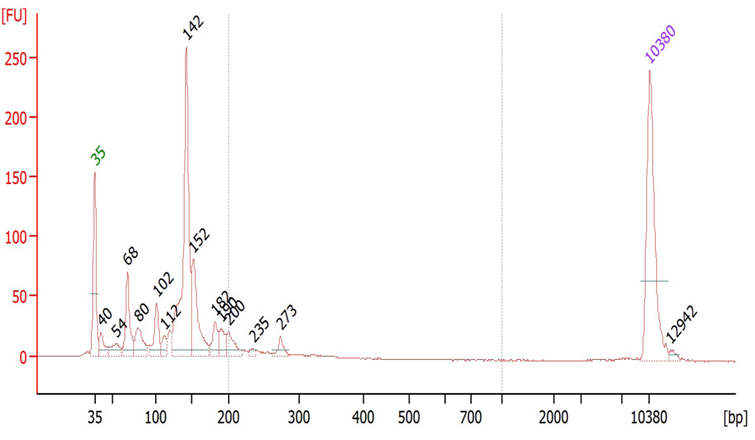
Suggested protocol for labeling Aminoallyl FLuc mRNA
This protocol is compatible for use with most water-soluble NHS esters*. If using a non-water soluble NHS-ester, different processing will be required. Precipitation in alcohol, such as isopropanol or ethanol, should be considered to remove excess NHS ester and any hydrolysis products. Multiple precipitations may be required.
*This protocol has been tested at small scales (100 ug) using Aminoallyl FLuc mRNA and Cyanine 5. Scales larger than 10 mg may require modifications to the protocol. The protocol has been designed to minimize mRNA degradation during the labeling process.
Salt exchange of Aminoallyl FLuc mRNA
1. Concentrate desired amount of Aminoallyl FLuc mRNA with 3K centrifugal filter. Reduce volume to ~0.25X of original.
2. Add 100 mM NaHCO3 to achieve total volume of 1.25X of original.
3. Repeat steps 1-2 at least three times. Add enough 100 mM NaHCO3 at final step to achieve 1X original volume of product. Concentration should be at ~ 1 mg/mL.
Labeling Reaction
4. Add 1X volume 100 mM NaHCO3 in H20. Mix gently by inverting several times.
5. Add 2X volume 2 mM NHS ester solution in DMSO. Mix gently by inverting several times.
6. Incubate 90 min at room temperature. If label is fluorescent, make sure to shield from light.
7. Add ~0. 35X volume 4M hydroxylamine in H20. Mix gently by inverting several times.
8. Incubate in dark for 15 min at room temperature. If label is fluorescent, make sure to shield from light.
Processing
9. Concentrate ~3X with 10K centrifugal filter device.
10. Add back 3X volume H2O.
11. Repeat steps 9-10 at least 6 times.